

Introduction to specification of SMILES and review of its effect of chemical toxicity prediction
-
摘要:
归纳了简化分子线性输入规范(simplified molecular input line entry specification, SMILES)编码规则, 探讨了分子结构输入及转化方法, 回顾了SMILES在毒性预测领域的适用性。SMILES规则能覆盖原子、离子、化学键、化合价、化学反应式的表达, 还能精准表达支链、环状、四面体、芳香族及手性结构, 同时涉及同分异构体和同位素的表达。SMILES能将几乎所有化学物的结构进行转换、输入, 并与大多数化学智能软件兼容, 结合TOPKAT和TEST软件获得的化学物毒性预测结果非常理想。SMILES是一种应用范围广、兼容性强的信息输入及搜索工具, 将会大力推动化学信息程序的发展。
-
关键词:
- 简化分子线性输入规范 /
- SMILES /
- 毒性预测 /
- 编码规则 /
- 化学信息程序
-
化学品的毒性问题关乎人身、财产和环境安全,还直接影响到生产、使用、经营、运输和仓储等环节的安全。因此,化学品的毒性评估是非常重要的安全性评价内容。然而,传统的毒理学研究以动物实验为基础,存在如周期长、成本高、灵敏度低等诸多弊端。此外,出于对动物福利和道德伦理的考虑,新型的化学物毒性预测方法亟待发展。近年来,随着计算机和信息科学的快速发展,人们开发了多款定量构效关系(quantitative structure-activity relationship,QSAR)软件[1-3],并已经在毒理学研究方面开展了广泛的应用。但是,如何将化学物分子结构输入至毒性预测软件,并与不同的毒性预测软件兼容是值得人们思考的问题。简化分子线性输入规范(simplified molecular input line entry specification,SMILES)为人们提供了一种有效而便捷的分子结构输入方法,还能很好地解决毒性软件兼容的问题[4-5]。由于SMILES基于字符输入,能方便快捷地将分子结构进行转化,生成智能化学软件兼容的语言,因此,它的使用能大力推动化学信息程序的发展,可应用于数据库的存取,作为研究者交换化学信息的平台,或作为人工智能语言、化学专家系统的一部分。本文拟通过介绍SMILES规范,并对其应用效果进行回顾,以推动SMILES编码在我国的应用和推广,推动我国化学品相关毒性预测软件的技术开发,以促进我国危险化学品安全管理和化工信息智能化的发展。
1. SMILES简介
20世纪80年代人们开发了化学信息系统,但并非人人都拥有化学结构作图软件,分子结构输入成为棘手问题。SMILES是一种利用ASC Ⅱ字符串明确描述分子三维结构和化学反应的规范,可用于解决将分子输入软件的问题[6]。最初由ARTHUR WEININGER和DAVID WEININGER于20世纪80年代末开发,并由日光化学信息系统有限公司(Daylight Chemical Information Systems Inc.)修改和扩展。
SMILES是一种非常有用的语言,仅需要简单的原子符号、键符号和语言规则就能完成分子结构的表达,并为其他化学智能软件提供化学物信息。这是因为SMILES基于字符,能提供一种方便快捷的分子结构输入及结构式直接转化的途径[7]。与其他著名的线性记法如威斯威瑟线性表达法(Wiswesser line notation,WLN)、线性排列的有机结构描述(representation of organic structure description arranged linearly,ROSDAL)、分子模拟线性表达法(SYBYL line notation,SLN)和国际化合物标识(international chemical identifier,InChI)等相比,SMILES被普遍认为是可读性最佳、应用性最广、兼容性最好的线性编码。
2. SMILES的基本规则
SMILES符号由一连串不间断的字母组成,采用纵向优先遍历树算法[8]将化学结构进行转化。在SMILES基本规则中,氢原子忽略不计,芳环结构采用链打开的方式或直接由凯库勒(Kekulé)形式表达。表达时,拆分键端的原子要用数字标记,支链写在小括号里。SMILES编码的五个要素分别为原子、化学键、支链、闭环和未连接。化学物的同分异构体遵循同分异构体(isomeric)SMILES编码规则。SMILES字符串可被大多数分子编辑软件导入,并转换成二维或三维的分子模型,以保证每个化学分子只有一个SMILES表达式[9]。
2.1 原子的SMILES表达
在SMILES表达式中,原子由相应的元素符号表示,即使用相应的英文字母;化学物的氢原子被省略;其他原子由相应的元素符号加中括号[]表达。由两个字母组成的原子,其元素符号的第二个英文字母要小写,如Cl、Br。当化学键明确且元素处于最低正常价态时,化学物可直接表达为元素符号,如B、C、N、O、P、S、F、Cl、Br和I,不用加中括号。芳香环类物质用小写字母c表达,而脂肪族用大写C表达。见表 1。
表 1 部分化学物的SMILES表达化学物名称 分子式 SMILES表达式 碳 C [C] 硫 S [S] 金 Au [Au] 甲烷 CH4 C 磷化氢 PH3 P 氨气 NH3 N 硫化氢 H2S S 水 H2O O 盐酸 HCl Cl 2.2 离子的SMILES表达
当元素的化合价高于最低正常价态,或存在于无机化学物中时,元素要加中括号表达。例如,元素硫[S]、元素金[Au]。与原子相连接的氢离子或其他任何离子都应被注明。连接的氢数量应采用字母H加数字来表达。正常电荷数的离子可采用元素符号加“+”或“-”加数量来表达,也可以直接采用多个“+”或“-”表示出所有电荷,如[Fe+3]与[Fe+++]是等效的表达。对于在中括号内的元素,如果没有标明电荷数,就认定电荷为零。部分离子的SMILES表达式见表 2。
表 2 部分离子的SMILES表达离子名称 SMILES表达式 质子 [H+] 二价铁离子 [Fe+2]或[Fe++] 羟基 [OH-] 碳酸氢根离子 [O=C(O)O-] 水合氢离子 [OH3+] 铵根离子 [NH4+] 钠离子 [Na+] 钙离子 [Ca+2] 氯离子 [Cl-] 硫离子 [S-2] 硫酸根离子 [OS(=O)(=O)O-2] 硝酸根离子 [ON(=O)O-] 2.3 化学键的SMILES表达
在SMILES编码规则中,单键、双键、三键和芳香族的离域键分别可由“—”“=”“#”和“:”符号来表达。相邻的原子由单键或芳香族的离域键连接时,通常无须符号表示(见表 3)。对于线性结构的物质,SMILES基于常规图解方法,并省略氢原子和单键。例如,6-羟基-1,4-己二烯(H2C=CH-CH2-CH=CH-CH2-OH)可采用三种等效的SMILES表达,分别为C=CCC=CCO、C=C-C-C=C-C-O、OCC=CCC=C。
表 3 部分线性化学物的SMILES表达化学物名称 分子式 SMILES表达式 乙烷 CH3CH3 CC 甲醛 CH2O C=O 乙烯 CH2=CH2 C=C 二氧化碳 CO2 O=C=O 二甲醚 CH3OCH3 COC 氰化氢 HCN C#N 乙醇 CH3CH2OH CCO 氢气 H2 [H][H] 苯 CH2=CHCH=CHCH=CH2 C:CC:CC:C 2.4 含支链化学物的SMILES表达
支链应该写在括号()内,并表达在所连接的主链原子之后。见表 4。
表 4 部分含支链化学物的SMILES表达化学物名称 三乙 异丁酸 3-丙基-4-异丙基-1-庚烯 结构式 


SMILES表达式 CCN(CC)CC CC(C)C(=O)O C=CC(CCC)C(C(C)C)CCC 2.5 环状结构的SMILES表达
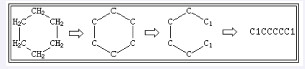
SMILES表达中,环状结构需断开一个化学键来表述分子结构。连接键可按任意顺序标注,键断裂处的原子后用阿拉伯数字1、2、3等标注。这里的数字只是用来标注闭环结构及闭环的数量。断裂后的环状化学物按上述线性化学物的三大原则进行表述。环己烷就是典型的例子,其SMILES表达为C1CCCCC1,见图 1。
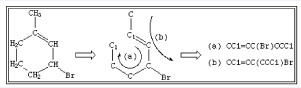
同一个化学物也可由多个不同的等效的SMILES表达式进行表述。例如,1-甲基-3-溴-环己烯可以用CC1=CC(Br)CCC1与CC1=CC(CCC1)Br两种等效的SMILES表达式来表达。见图 2。
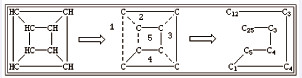
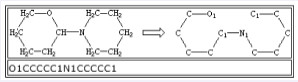
有些情况下,一个原子可能会涉及多个闭环结构,例如立方烷,其SMILES表达式应该为C12C3C4C1C5C4C3C25(见图 3)。其中的1、2、3、4、5说明共有5个闭环结构,数字是每个闭环结构断裂处的位置标志。对于各自独立的闭环结构,有时断裂键处标注的数字可以重复使用,如图 4所示结构的SMILES表达式为O1CCCCC1N1CCCCC1。键断裂位置的选择没有强制性要求,可选择闭环结构中任意一个单键进行断裂,在键断裂的相邻两个碳原子上标注阿拉伯数字,标注有数字的相邻碳原子是单键连接的,整体构成闭环结构。
键断裂处重复使用相同的数字标注是为了满足表达10个或10个以上的闭环结构。化学物结构中含有10个以上闭环的情况是极为罕见的。必要时可采用两位数字表达并加百分号“%”连接。例如,C2%13%24指一个碳原子同时具有闭环2、13、24。2、13、24说明该分子结构非常复杂且庞大,可能拥有24个闭环结构,其有一个碳原子同时参与3个闭环结构,分别是第2、第13和第24个闭环结构,且这3个闭环结构都是在与此碳原子相连键的3处键断裂处进行表达的。
2.6 未连接结构的SMILES表达
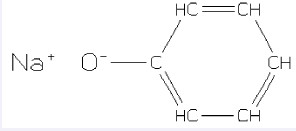
未连接结构化学物采用点符号“.”来隔开每个单独的结构。离子或配体可按任意的顺序标注出。一个电荷无须与另一个电荷组成配体,化学物也无须净电荷为零。有时,SMILES可表达一个离子包含于另一个离子化学物中。如苯酚钠,其SMILES表达式为[Na+].[O-]c1ccccc1或c1cc([O-].[Na+])ccc1。其结构式见图 5。
2.7 连接结构的SMILES表达
原子后面加数字表示这些原子是直接相连接的。连接键可能是很明确地用键符号或者闭环数字来表达的,也可能是不明确方向的芳香族键。以点符号“.”隔开的相邻原子表示原子间不是相互连接的,即使原子属于同一个连接的化学物。如C1.C1指CC(乙烷)分子。而表 3中乙烷的表达式CC是简化表达式。
2.8 同分异构体的SMILES表达
SMILES也能表述同位素、双键结构和手性分子。所有的同分异构体在SMILES规则中都是具有选择性的,允许手性分子的任意结构。这点不同于其他的化学语言,如卡恩-英格尔-普雷洛格规则(Cahn-Ingold-Prelog,CIP)和国际纯粹与应用化学联合会(International Union of Pure and Applied Chemistry,IUPAC)命名法规则要求严格的手性分子说明。SMILES规则中同分异构体的多重选择性在计算机指令、结构检索、模式匹配等操作中具有明显优势。目前,全球化学物的结构信息、手性中心并非都是已知的、被研究或已报道的。因此,SMILES规则在同分异构体中的应用具有深远的意义。
2.9 同位素的SMILES表达
同位素的表达采用元素符号前加原子量在中括号内标注出。见表 5。
表 5 碳同位素的SMILES表达元素或化学物名称 SMILES表达式 碳-12 [12C] 碳-13 [13C] 碳 [C] C-13甲烷 [13CH4] 2.10 双键构型的SMILES表达
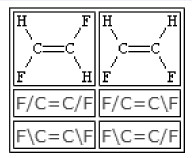
双键周围的构形可用符号“/”和“\”来表达单键或芳香键的方向。当原子间存在双键时,“/”和“\”表明原子间连接的相对方向。E-1,2-二氟乙烯和Z-1,2-二氟乙烯的SMILES表达式如图 6所示。SMILES不同于传统的CIP语言,SMILES可采用局部手性表达,并非绝对手性,允许局部说明的分子构型。见表 6。
表 6 某双键分子构型的SMILES表达完全手性说明 局部手性说明 结构式 

SMILES表达式 F/C=C/C=C/C F/C=C/C=CC “局部手性”源于对“局部对称性”的一般讨论,是个与对称性相关的概念。通俗地说,出于分子的对称中心或对称面上的点都是非局部手性的,其他所有的部分都是局部手性的,也称它们处于手性环境。手性分子的各部分自然都是局部手性的,非手性分子各部分却不都是非局部手性的。例如,甲烷CH4的五个原子都是非局部手性的,但不在对称面上的所有空间则是局部手性的。1S,2R-1,2-二氯-1,2-二溴乙烷是非手性的,但它所有原子都是局部手性的。
“绝对手性”的概念与“局部手性”有所不同。当连接到中心碳原子上的a、b、c、d是不同基团时,分子是绝对手性结构。假设分子中四个取代基按卡恩-英格尔-普雷洛格规则、CIP规则以a > b > c > d的顺序排列,如果从中心碳原子到最小的基团d方向,观察到a→b→c是顺时针方向,则这个碳中心的构型被定义为R;否则就认定为S。以这个规则来观察前述的乳酸、丙氨酸、甘油醛,不难看出它们的绝对构型如下:D型的甘油醛和乳酸为R构型,天然的氨基酸如丙氨酸则是S构型。
2.11 四面体构型的SMILES表达
SMILES采用基于局部手性的通用手性表达方式,不将手性中心周围的相邻原子进行编码,而是按相邻原子间的顺序来定义方向。SMILES能接受任意的原子排列顺序,当分子结构发生改变或重组时,仍能保留手性分子的特征。四面体是最常见、最简单的手性结构。四个相邻的原子围绕着同一个原子排列,该原子为“手性中心”。如果四个相邻的原子互不相同,那么其镜像结构是不同的,两个相对应的镜像体作为手性中心的两个异构体。SMILES规则中,四面体中心采用元素符号加“@”或“@@”来简化手性说明。如果手性中心未作说明,那么手性分子的手性结构是未明确的。
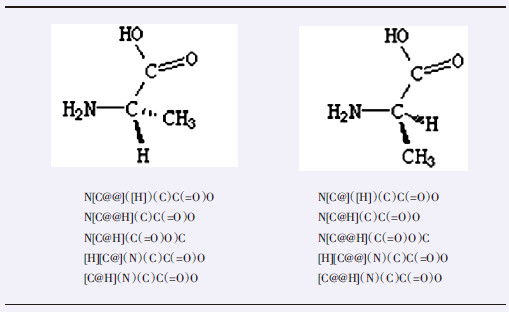
如表 7“明确手性结构”一栏第一行的SMILES表达式N[C@](C)(F)C(=O)O(即依次为氨基、中心碳、甲基、氟、羧基基团),从氨基N开始看到手性中心C,另外三个相邻的原子是按逆时针方向来描述的。第二行SMILES描述是按顺时针方向表达:N[C@@](F)(C)C(=O)O。符号“@”指相邻基团绕中心原子呈逆时针顺序排列。符号“@@”指相邻基团绕中心原子呈顺时针顺序排列。
表 7 某些手性结构的SMILES表达未明确手性结构 明确手性结构 结构式 

SMILES表达 NC(C)(F)C(=O)O或NC(F)(C)C(=O)O N[C@](C)(F)C(=O)O或N[C@@](F)(C)C(=O)O 如果中心碳原子是SMILES表达中的起始原子,并带有氢原子,此时,该碳中心原子仍具有手性特征,其所带氢原子可作为四个相邻基团之一,围绕中心碳原子组成四面体。此中心碳原子内带的氢原子记作[H]。丙氨酸几种有效的SMILES表达式见图 7。
3. 一般的手性规则
手性化学物要比四面体结构的物质多得多。上述用“@”符号表达四面体是普通手性有序排列结构的一种特殊情况。SMILES规则中的手性说明分为3个部分:“@”符号、双字母的手性划分标志、数字化的手性排列标志。默认的手性阶数是指连接数;原子连接数为4的物质被默认为是四面体(记作TH)构造。大多数的手性结构都有两种以上的选择可能。大多数情况下,“@1”指SMILES表达中原子按轴向逆时针顺序排列,“@2”则指SMILES表达中原子按轴向顺时针顺序排列。
SMILES能处理手性规范中的所有情况,包括当分子结构对称时异构体数量的减少,因为对称替代使中心原子成为非手性中心。SMILES语言考虑到了已知结构和未明确结构的情况。对于任何给定的结构,SMILES系统都能产生唯一对应的异构体表达式,对所用的手性分子结构进行准确识别,SMILES还能处理更多复杂的结构,如四面体、丙二烯结构、正方形平面、三角-双锥体、八面体,等等。
4. SMILES中其他规则
4.1 氢的规则
通常情况下,有机物结构中的氢原子无须被标注出来。氢包括三种表达方式:1)氢原子以普通单键相连,处于正常的化学价态时,无须标注氢原子;2)中括号中的氢原子,并加数字表明氢的数量;3)[H]指氢原子。
SMILES语言对有机物和无机物不存在差异性。任意原子上连接的氢原子数量可以不作具体说明。例如,丙烷可输入为[CH3][CH2][CH3]或CCC(采用SMILES简化表达式CCC常见)。以下四种情况,氢需要明确的说明:1)带电荷的氢,如一个质子[H+];2)氢原子与氢原子相连接,如氢气分子[H][H];3)氢原子不止与另一个原子相连接,如桥连氢;4)同位素氢原子,如重水[2H]O[2H]。
4.2 芳香族化学物的规则
基于分子对称性特征,SMILES能推导出芳香族结构的明确化学命名法则。如果使用者更偏爱脂肪族的凯库勒结构,那么在表达芳香族有效结构时可以不输入芳香族结构。芳香族结构也可以直接采用小写字母的原子符号来表达,如n指芳香族氮化合物的氮原子,这样能简化明确的化学结构说明,也更接近于分子模型的真实结构,因而被广泛使用。SMILES规则采用休克尔(Hueckel’s)规则的扩增版来定义芳香族分子和离子,还能为芳香族提供带电荷的、杂环的和电荷不平衡的环状结构的表达。
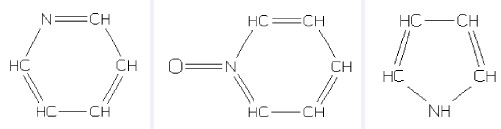
吡啶、吡啶-N-氧化物、吡咯是三种常见的芳香族氮化合物(结构见图 8),SMILES表达式分别为n1ccccc1、O=n1ccccc1、[nH]1cccc1。值得注意的是吡咯有三种等效的SMILES表达式:[nH]1cccc1、[H]n1cccc1、N1C=CC=C1。
4.3 化合价的规则
SMILES使用分子结构模型时不对化合价做明确定义,这也是SMILES的一大优势,针对一个分子结构模型,SMILES能描述出不同的化合价。例如,硝基甲烷可表达为CN(=O)=O或者带电表达式C[N+](=O)[O-]。两种表达方式都是正确的,但我们更倾向于采用后一种表达方式,因为当分子呈非对称结构时,采用电荷分离结构表达能避免原子处于非正常化合价态时引起的歧义。例如,重氮甲烷表达为C=[N=]=[N-]时优于用C=[N]=[N]表达。
5. SMILES规范在化学物毒性预测中的应用回顾
SMILES所表达的分子结构式能为其他化学智能软件提供化学物结构信息,与毒性预测软件ECOSAR、DEREK、TOXTREE、ACD/Labs、TEST、TOPKAT等兼容,应用于毒性预测领域。实际应用中,SMILES能快速将待测化学物结构转换成计算机能有效识别的符号输入,成功解决了智能软件兼容的问题。尽管国际纯粹与应用化学联合会推荐国际化学物标识(InChI)作为标准的分子表达式,但是迄今为止,SMILES是应用最为广泛、兼容性最好的简化线性输入编码。SMILES编码是近年来国际上化学物质结果信息表示的新型技术和标准,已经成为国际上通用的统一标准。将SMILES编码作为化学物质结构信息保存在数据库中,既减少占有系统空间,提高服务器检索效率,更能体现化学物质属性信息,便于专业人员进行查询分析。相比于其他规则,SMILES规则具有简单便捷、兼容性强、适用范围广、精准高效等特点。SMILES还可广泛应用在化学物数据库中结构的计算机化搜索、分子二维图片结构转换、分子三维模型转换等方面,还可作为智能化工软件开发的基础工具。我们曾尝试采用SMILES结合TEST[10]、TOPKAT[11-13]软件对321种化学物的健康毒性进行预测,并将预测值与文献值进行比较。结果(见表 8)发现,SMILES与两款毒性预测软件相容性良好,依据SMILES的分子结构表达毒性预测结果与文献值很接近,预测值与文献值的线性决定系数R2为0.854 ~ 0.986(P < 0.01),毒性预测的正确率约85%[14],表明SMILES的适用性很强。
表 8 SMILES在化学物毒性预测中的应用化学物名称 SMILES表达式 LD50/(mg/kg)(文献值) LD50/(mg/kg)(采用TOPKAT软件获得的预测值) LD50/(mg/kg)(采用TEST软件获得的预测值) 对羟基苯甲腈 c1cc(O)ccc1C#N 450 370.2 765.41 1-萘酚 c12cccc(O)c1C=CC=C2 1 870 2 000 1 564.72 甲苯二异氰酸酯 c1cc(C)c(N=C=O)cc1N=C=O 5 110 3 600 4 164.35 邻硝基苯胺 c1cccc(N)c1N(=O)=O 1600 729.2 1 294.68 六亚甲基二异氰酸酯 O=C=NCCCCCCN=C=O 746 963.4 1 367.45 苄硫醇 c1ccccc1CS 493 582.1 476.37 苯乙酸乙酯 c1ccccc1CC(=O)OCC 3 300 2 100 2 722.03 6. 小结
本文系统介绍了SMILES编码规则,并对其应用于化学物毒性预测领域的效果进行了回顾,发现SMILES能覆盖所有的化学结构,与多款化学智能软件兼容性优良,获得的预测结果也很理想。SMILES编码规则已被很多国际领先的化学品研究管理机构、化学品制造商采用。然而,我国目前还没有化学物质数据库采用这一方法,该方法亟待在我国应用和推广。目前,SMARTS作为SMILES的改进版,增加了允许使用通配符表示原子和化学键的规则。于是,SMILES可被应用在化学物数据库中化学结构的信息搜索。今后,SMILES的使用能大力推动智能化学信息程序的发展。
志谢: 本项研究工作得到了上海市科委上海化学品公共安全工程技术研究中心项目的资助 -
表 1 部分化学物的SMILES表达
化学物名称 分子式 SMILES表达式 碳 C [C] 硫 S [S] 金 Au [Au] 甲烷 CH4 C 磷化氢 PH3 P 氨气 NH3 N 硫化氢 H2S S 水 H2O O 盐酸 HCl Cl  下载: 导出CSV
下载: 导出CSV
表 2 部分离子的SMILES表达
离子名称 SMILES表达式 质子 [H+] 二价铁离子 [Fe+2]或[Fe++] 羟基 [OH-] 碳酸氢根离子 [O=C(O)O-] 水合氢离子 [OH3+] 铵根离子 [NH4+] 钠离子 [Na+] 钙离子 [Ca+2] 氯离子 [Cl-] 硫离子 [S-2] 硫酸根离子 [OS(=O)(=O)O-2] 硝酸根离子 [ON(=O)O-]
下载: 导出CSV
表 3 部分线性化学物的SMILES表达
化学物名称 分子式 SMILES表达式 乙烷 CH3CH3 CC 甲醛 CH2O C=O 乙烯 CH2=CH2 C=C 二氧化碳 CO2 O=C=O 二甲醚 CH3OCH3 COC 氰化氢 HCN C#N 乙醇 CH3CH2OH CCO 氢气 H2 [H][H] 苯 CH2=CHCH=CHCH=CH2 C:CC:CC:C
下载: 导出CSV
表 4 部分含支链化学物的SMILES表达
化学物名称 三乙 异丁酸 3-丙基-4-异丙基-1-庚烯 结构式 SMILES表达式 CCN(CC)CC CC(C)C(=O)O C=CC(CCC)C(C(C)C)CCC
下载: 导出CSV
表 7 某些手性结构的SMILES表达
未明确手性结构 明确手性结构 结构式 SMILES表达 NC(C)(F)C(=O)O或NC(F)(C)C(=O)O N[C@](C)(F)C(=O)O或N[C@@](F)(C)C(=O)O
下载: 导出CSV
表 8 SMILES在化学物毒性预测中的应用
化学物名称 SMILES表达式 LD50/(mg/kg)(文献值) LD50/(mg/kg)(采用TOPKAT软件获得的预测值) LD50/(mg/kg)(采用TEST软件获得的预测值) 对羟基苯甲腈 c1cc(O)ccc1C#N 450 370.2 765.41 1-萘酚 c12cccc(O)c1C=CC=C2 1 870 2 000 1 564.72 甲苯二异氰酸酯 c1cc(C)c(N=C=O)cc1N=C=O 5 110 3 600 4 164.35 邻硝基苯胺 c1cccc(N)c1N(=O)=O 1600 729.2 1 294.68 六亚甲基二异氰酸酯 O=C=NCCCCCCN=C=O 746 963.4 1 367.45 苄硫醇 c1ccccc1CS 493 582.1 476.37 苯乙酸乙酯 c1ccccc1CC(=O)OCC 3 300 2 100 2 722.03
下载: 导出CSV
-
[1] MYINT K Z, WANG L, TONG Q, et al. Molecular fingerprint-based artificial neural networks QSAR for ligand biological activity predictions[J]. Mol Pharmaceutics, 2012, 9(10):2912-2923. doi: 10.1021/mp300237z
[2] WANG M J, ZHAO X B, WU D, et al. Design, synthesis, crystal structure, insecticidal activi-ty, molecular docking, and QSAR studies of novel n3-substituted imidacloprid derivati-ves[J]. J Agric Food Chem, 2014, 62(24):5429-5442. doi: 10.1021/jf501108j
[3] CHERKASOV A, MURATOV E N, FOURCH ES D, et al. QSAR Modeling:where have you been? where are you going to?[J]. J Med Chem, 2014, 57(12):4977-5010. doi: 10.1021/jm4004285
[4] KARWATH A, DE RAEDT L. SMIREP:predicting chemical activity from SMILES[J]. J Chem Inf Model, 2006, 46(6):2432-2444. doi: 10.1021/ci060159g
[5] BONE R G A, FIRTH M A, SYKES R A. SMILES extensions for pattern matching and molecular transformations:applications in chemoinformatics[J]. J Chem Inf Comput Sci, 1999, 39(5):846-860. doi: 10.1021/ci990422w
[6] ANDERSON E, VEITH G D, WEININGER D. SMILES: a line notation and computerized interpreter for chemical structures. Report No. EPA/600/M-87/021[R]. Duluth, MN 55804: U.S. EPA, Environmental Research Laboratory-Duluth, 1987.
[7] WEININGER D, WEININGER A, WEININGER J L. SMILES. 2. algorithm for generation of unique SMILES notation[J]. J Chem Inf Model, 1989, 29(2):97. doi: 10.1021/ci00062a008
[8] HELSON H E. Structure diagram genera-tion[J]. Rev Comput Chem, 2007, 13:313-398.
[9] WEININGER D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules[J]. J Chem Inf Comput Sci, 1988, 28(1):31-36. doi: 10.1021/ci00057a005
[10] US Environmental Protection Agency. User's Guide for T.E.S.T. (version 4.0) (Toxicity Estimation Software Tool)[R]. 2011.
[11] BHHATARAI B, WILSON D M, PARKS A K, et al. Evaluation of TOPKAT, Toxtree, and derek nexus in silico models for ocular irritation and development of a knowledge-based framework to improve the prediction of severe irritation[J]. Chem Res Toxicol, 2016, 29(5):810-822. doi: 10.1021/acs.chemrestox.5b00531
[12] VENKATAPATHY R, MOUDGAL C J, BRUCE R M. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction[J]. J Chem Inf Comput Sci, 2004, 44(5):1623-1629. doi: 10.1021/ci049903s
[13] 黄晓龙.国外化学物结构-毒性数据库"TOPKAT"简介[J].中国新药杂志, 2005, 14(3):262-264. doi: 10.3321/j.issn:1003-3734.2005.03.003 [14] 王思怿, 范宾. TOPKAT和TEST软件在化学物毒性预测中的应用[J].职业卫生与应急救援, 2017, 35(1):1-5. http://zyws.cbpt.cnki.net/WKD/WebPublication/paperDigest.aspx?paperID=890cbd95-4482-49c7-ae53-77fd38d11cd5 -
期刊类型引用(0)
其他类型引用(3)
计量
- 文章访问数: 1034
- HTML全文浏览量: 163
- PDF下载量: 34
- 被引次数: 3
